Modeling the Fullerene Distribution in BHJ Solar Cells
James T. Rogers
CHE210D Spring 2009 Final Project
Summary
A simple model was used to study the effect of polymer molecular weight on the distribution of fullerene molecules within a bulk heterojunction (BHJ) solar cell. Measurements of the pair distribution function (PDF) derived from this model indicate improved local structural ordering of the fullerene molecules with increasing molecular weight. This is well correlated with recent experimental results which have shown drastic increases in solar cell performance with increasing polymer molecular weight. Further development of this model should allow identification of polymer/fullerene chemistries which may result in even more optimal blend morphologies.
Background
High efficiency bulk heterojunction (BHJ) “plastic” solar cells are based on phase-separated blends of polymer semiconductors and fullerene derivatives. As a result of nanometer scale phase separation, excitons formed after absorption of solar irradiation need to diffuse only a short distance to heterojunctions where charge separation occurs. Although significant advancements in the performance of polymer-based photovoltaic devices have been made in recent years by rationally designing polymers which absorb at optimal wavelengths of the solar spectrum, the ability to systematically control the morphology of the phase separated and bicontinuous donor/acceptor network remains a critical unmet challenge to optimizing the efficiency of organic solar cells.
Simulation methods
The degree of “Phase-separation” in the BHJ system was measured by simulating a two component mixture of polymer and fullerene. Fullerenes were modeled as spheres interacting through a Lennard-Jones potential. Polymers were modeled as a Lennard-Jones chains which possess both “bonded” monomers (interacting through a harmonic potential) and non-bonded monomers (interacting through a second Lennard-Jones potential). Lennard-Jones parameters (σ and ε) were chosen to reflect the relative length scales and interaction strengths between the components, respectively. Pair distribution functions (PDFs) were then calculated by sampling fullerene-fullerene distances in the equilibrated mixture at periodic time intervals.
The molecular dynamics simulation was carried out in the microcanonical ensemble using the velocity Verlet algorithm. Prior to the production run from which the PDFs were measured, the system underwent two equilibration periods. For numerical stability an initial equilibration period was carried out using velocity rescaling, this was followed by a second equilibration period of equal length during which time the average total energy was recorded. Just prior to the production run, the average total energy was compared to the potential energy of the system, and used to rescale the molecule velocities in order to avoid the need for periodic rescaling during the production run.
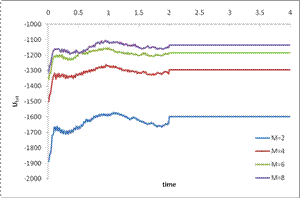
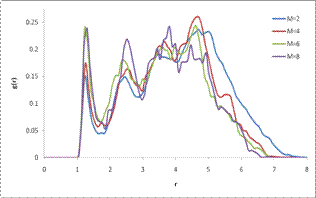
Results and interpretation
The results of the simulations shown above indicate a large increase in the average total energy of the mixture with increasing polymer molecular weight. Since both the density and the polymer:fullerene ratio were kept constant between simulations this increase can be attributed to increased packing frustration of the polymer chains with increasing molecular weight. More importantly, the simulations illustrate that fullerene molecules become more tightly correlated as the molecular weight of the polymer component increases.
Examination of the system dynamics reveals one possible mechanism for the observed improvement in structural ordering of the fullerene molecules. It can be observed that as the molecular weight increases the polymers become more entangled in the matrix which limits their ability to diffuse and allow fullerene molecules into the matrix. Although this simple model has yielded some nice results, current deficiencies in the model arise from two sources. First, the precise values of the interaction energy between like and dislike components is unknown, thus only approximate interaction strengths can be entered into the model. The more serious deficiency of the model is the assumption of Lennard-Jones chain behavior of the polymer. In actuality, due to the conjugated nature of the polymers involved in BHJ systems, chains are much stiffer and a bending potential should be included.
Movie
The accompanying movie is a valuable tool for visualizing the mechanism of improvement in fullerene-fullerene ordering. In the video, blue molecules represent the polymer chains and red molecules represent the slightly smaller fullerene molecules. The MD simulation illustrates how fullerene molecules, once separated into one region of the mixture, have great difficulty diffusing into the matrix as a result of the low mobility of the long polymer chains.