A Coarse-grained MD Simulation of Micellization Process
Mansi Seth
CHE210D Spring 2009 Final Project
Summary
A Molecular Dynamics Simulation was performed to study the process of Micellization of surfactant molecules in water, using an implicit solvent, coarse-grained model. The Critical Micelle Concentration (C.M.C) was determined for an ionic surfactant. The Effect of Temperature on C.M.C was also studied. Micellization is observed only above a certain critical concentration, after which there is an increase in concentration of micelles, with increase in surfactant concentration. An increase in temperature leads to increase the C.M.C of the surfactant. The results obtained are thus, in qualitative agreement with experimental observations and those from other simulations of ionic surfactant molecules.
Background
Amphiphilic molecules spontaneously self-associate in aqueous solutions, readily forming aggregates such as micelles, bilayers and vesicles, above the critical micelle concentration (c.m.c). Atomistic MD simulation of such a system is difficult since the equilibration timescales are very large. In this study, a coarse- grained model is used which is computationally faster to carry out. Such a model can be used to verify the results obtained in experiments. It can also be used to predict the change in c.m.c with temperature.
Simulation methods
In this study, an implicit-solvent, coarse-grained model is used to describe the interactions between the surfactant molecules. There are two types of particles, headgroup particles (HG), and tail particles (T). Each monomer consists of 6 units: 1 HG unit followed by 5 T units. The solvent (water) is accounted for in an implicit manner, by considering an effective interaction between the HG units. The surfactant considered is thus, an ionic surfactant.
The interaction between the HG - HG units is modeled to be completely repulsive, comprising of only the repulsive part of the Lennard-Jones (LJ) Potential. Similarly, the interaction between the HG-T units is also completely repulsive. These are given as follows:
UHG-HG (r) = 4 εHG-HG (r/σHG-HG)-12 ………………………………………… (1)
UT-HG (r) = 4 εHG-T (r/σHG-T)-12 ……...…………………………………….. (2)
The bonded tail units interact via a harmonic potential given by:
UT-T(bonded) (r) = 0. 5 k (r - ro)2 ……………………………………….......(3)
Finally, the non-bonded tail units interact via the LJ potential:
UT-T (non-bonded) (r) = 4 εT-T { (r/σHG-T)-12 - (r/σHG-T)-6 }………………(4)
Thus, the total Interaction Potential is given as:
UTotal (r) = UHG-HG (r) + UT-HG (r) + UT-T(bonded) (r) + UT-T (non-bonded) (r)
The hydrophobic interactions between the terminal tail units are made stronger, so as to favour formation of spherical micelles. The various interaction parameters are given in Table 1.0
Table 1.0: Interaction parameters of LJ potential
|
Unit Types |
є (kJ/mol) |
σ (nm) |
K (N/m) , ro (nm) |
|
HG-HG |
1.25 |
0.6 |
- |
|
T-T (non-bonded) |
0.5 |
0.4 |
- |
|
HG –T(non-bonded) |
2.5 |
0.5 |
- |
|
T-T(bonded) |
- |
- |
1000, 0.15 |
The simulation is performed using the Velocity Verlet Algorithm for Molecular Dynamics. The HG and T particles are initially placed on a Cubic lattice. The particles are given random, initial velocities. MD simulations are then performed for 300,000 timesteps, with Δt = 0.001 τ, where τ is the characteristic time scale (mσ2T-T/єT-T). With the parameters given in Table 1.0, τ = 1.5ps. In order to obtain constant NVT conditions, the system is first equilibrated to the desired temperature using velocity rescaling. Of the 300,000 timesteps, the first 200,000 steps are used for equilibration. The simulation was carried out 210 particles, thus having 35 monomers of the surfactant.
In order to determine whether a particular monomer is part of a micelle, or in effect, to determine the aggregation number, a criterion based on interatomic distance was used. The distance between the terminal tail units was computed and all pairs (i, j) for which rij < dc, were assumed to be in the same micelle, where dc is the chosen, threshold separation. The Total Energy of the system was observed to determine equilibrium. The Average Potential Energy was monitored to determine micelle formation.
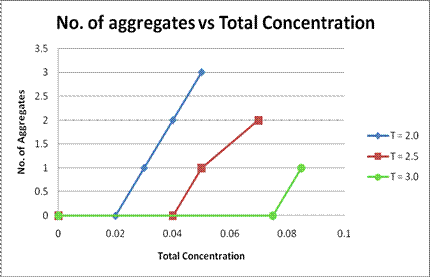
Figure 1. Number of Aggregates vs Total Concentration of Surfactant at different temperatures.
Results and interpretation
It was found that micelles are formed only above a certain density, or concentration. This is the c.m.c of the surfactant. On increasing the density above the c.m.c, the number of aggregates increased, as seen in Figure 1. However, at higher densities, the aggregation number also increased, leading to the formation of a single, larger micelle. On increasing the Temperature the c.m.c was found to increase as shown in Figure 2. The variation of the average potential energy with temperature, at a fixed density is shown in Figure 3. It is found that at a constant density, as the temperature decreases, the potential energy also decreases, and after a particular temperature, it remains almost constant. This temperature corresponds to the temperature at which micelles being to form, at that density.
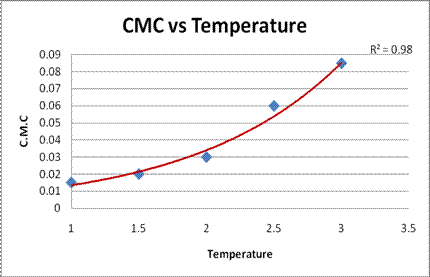
Figure 2. Effect of Temperature on the Critical Micelle Concentration: Blue squares are simulation values and the red line is an exponential fit to the points.
![]()
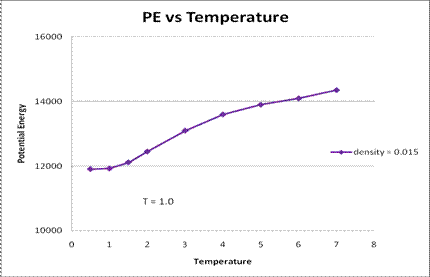
Figure 3. Potential Energy vs Temperature for a constant density of 0.015 (averaged over 5 simulation runs) . The error bars are smaller than the marker size.
It can be concluded that the c.m.c increases in an exponential manner with an increase in Temperature. At a constant density, there is a fixed temperature at which micelles begin to form. Thus at higher temperatures, the surfactant prefers to stay in the monomeric state, until the concentration is high enough, wherein it becomes energetically favorable for them to form micelles.
The model used has several deficiencies. For one, it does not include an angle bending potential between the bonded tail particles, and thus the tails are more flexible than they actually are. Furthermore, the size of the system is quite small. It would be appropriate to have a larger system size, in order that the results may be in quantitative agreement with experiments. For example, it is not possible to obtain correct aggregation numbers, since there is a limitation of small number of atoms. Another deficiency is that the effect of increase in temperature on the headgroup area, and thus the hydration interactions, is not accounted for. If this were taken into account, the c.m.c would show a minimum in the graph of c.m.c vs Temperature, wherein the c.m.c would initially decrease, and then increase with Temperature.
The model can thus be improved by accounting for the above mentioned deficiencies.
Movie
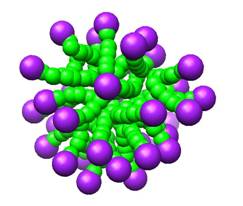
The movie shows the formation of a single micelle, with an aggregation number of 35, at a temperature of 1.5, and density of 0.25, which is above the c.m.c value of 0.02 for that temperature.
Source code
References
- Arben Jusufi, Antti-Pekka Hynninen, and Athanassios Z. Panagiotopoulos, “Implicit Solvent Models for Micellization of Ionic Surfactants,” The Journal of Physical Chemistry B 112, no. 44 (November 6, 2008): 13783-13792, doi:10.1021/jp8043225.
2. Computer simulations of a water/oil interface in the presence of micelles
B. Smit, P. A. J. Hilbers, K. Esselink, L. A. M. Rupert, N. M. van Os, A. G. Schlijper
Nature 348, 624-625 (13 December 1990)