Gallery of final projects
Students in the course designed and performed simulations of
coarse-grained models for a variety of systems of interest to them.
As a part of their projects, students developed movies of
simulation trajectories to visualize their results. The
titles below are links to the detailed web pages for each project,
where one can find the full results, analyses, and source
code. The associated images below also link
to movies. |
|
|
Polyelectrolyte
Complexation
Debbie
Audus
Two oppositely charged polymers are studied using molecular dynamics.
At high enough electrostatic strengths, these polymer form complexes.
The radius of gyration squared of these complexes is calculated as a
function of polymer length (N) and electrostatic strength
(λ). The resulting trends are the effect of various
interacting driving forces. |
|

|
The
Hydrophobic
Interaction in a
Spherically-Symmetric Water Bath
Aviel
Chaimovich
The hydrophobic interaction (HI) is perhaps the most significant
driving-force in living organisms. In this work, I
demonstrate a fundamental particularity of the HI: the notion that it
is mostly caused by the medium. I employ a Monte Carlo
simulation to achieve this task. |
|

|
The
Radius of Gyration for Charged Polymers
Peter
Chung
In random coil behavior, there is a power-law relationship between the
radius of gyration and the polymer length: Rg = R0 Nν.
Theoretically and experimentally, it has been shown that for
uncharged polymers ν ½. Recent experimental
evidence has shown that the radius of gyration for charged chemically
unfolded proteins has a power-law exponent of ν = .605
± .0271. A Monte-Carlo simulation of a linear polymer with a
Lennard-Jones and screened Coulomb potential was done to confirm the
experimental results. |
|

|
Nucleation
of Hard Sphere Crystals
Nathan
Duff
Hard spheres provide a model of colloid suspensions. Nucleation of hard
spheres from a metastable liquid to a crystal was studied. The free
energy, ΔG(n) of solid clusters was found to decrease with
increasing pressure. Finite size effects for the studied system size
(500 spheres) allow for spontaneous crystallization which does not
occur in the larger system (3375 spheres) studied by Auer and Frenkel.1
The results suggest a decreased barrier to nucleation due to finite
size effects. |
|
|
Shear
Flow Simulation: Velocity Field, Flow Boundary Conditions and Flow
Structure
Chia-Chun
Fu
Molecular dynamics simulations are carried out to investigate
Lennard-Jones liquids sheared between two solid walls. Velocity
profiles, flow boundary conditions and flow structures were studied for
various wall-fluid interactions. For weak wall-fluid interactions, the
flow structure is less ordered and slip was observed. For larger
wall-fluid interactions, the flow structure is more ordered and the
first one or two fluid layers near the walls were moving with the wall.
|
|
|
Capacitor
Charging Dynamics of Dilute Soft-Sphere Ions in an Implicit Solvent
Brian
Giera
A dilute system of equally sized cations and anions suspended in an
implicit solvent between two capacitor plates was simulated.
Concentration profiles were generated as a function of time and the
behavior of double layer formation was observed after the plates were
charged. This model can be used to investigate double layer formation,
which is a critical mechanism of supercapacitor charging.
|
|
|
Cluster
Formation in
Charged
Particles
David
Hassan
In this project I studied how the formation of Lennard-Jones clusters
was effected by electrostatic repulsion. In particular, I wanted to
test if the so called “magic cluster numbers” were
affected by the presence of a Coulomb interaction. The problem was
studied using a MD simulation, and the amount of charge present was
varied over the simulations. |
|

|
Scaling
law in thickness of polymeric brushes
Su-Mi
Hur
Here I study how polymer brush layer thickness changes depending on the
polymer chain length N and grafting density ρ is studied
through MD simulation. Each polymer chain is modeled as beads
and springs with shifted LJ and harmonic potentials. An additional
harmonic potential between grafting point and first bead in the chain,
as well as, a wall potential that interacts with all monomers is
introduced. Obtained results shows that high of brush layer h increases
linearly as N increases. h does not depend on ρ for
small values of ρ. However, h increases as ρ increases
for higher values of ρ and observed scaling follows
theoretical prediction. |
|
|
Clustering
of sodium chloride in implicit water solvent
Brandon
Knott
Simulation of nucleation from solution in condensed phases at constant
chemical potential can be achieved by utilizing an accurate effective
potential with implicit solvent. The distribution of cluster
sizes in a metastable system is an important parameter for nucleation,
as this process is driven by microscopic fluctuations in
density. Here I present grand canonical Monte Carlo results
showing that increasing the chemical potential in a system of chloride
and sodium ions shifts the cluster size distribution towards larger
clusters, a trend that makes a fluctuation large enough to form a
critical-sized nucleus more likely. |
|
|
Radius
of gyration of a polymer
Edmund
Lin
The radius of gyration is often used to characterize the dynamic
trajectory of flexible systems for molecular systems. Here, a small
radius of gyration indicates the polymer is relatively compact, meaning
throughout its trajectory the polymer spends most of its time as a
folded structure. We can measure the distribution of the radius of
gyration of a polymer’s dynamic trajectory to characterize
folding patterns. |
|

|
Liquid-Solid
Phase
Transition in
Lennard-Jones Particles
Michael
A.
Lovette
The liquid-solid transition in Lennard-Jones particles was investigated
through a series of Monte Carlo simulations in the liquid-solid
coexistence region. These simulations were stitched together
using WHAM and the phase boundary between the liquid-solid coexistence
region and the solid region of the phase diagram was observed. |
|

|
Phase
Separation in Block
Copolymer Melts
Zoltan
Mester
Molecular dynamics simulations are carried out on polymer chains made
up of all A or B monomers with hydrogen bonding head groups, and phase
segregation is characterized via radial distribution functions. The
radial distribution functions confirm that phase segregation is
stronger at lower temperatures, with nearly complete phase segregation
at T=1.2. At high temperature (T=2), the radial distribution functions
are qualitatively similar to that of a mix of Lennard-Jones particles
in the liquid phase that do not phase separate, suggesting low
segregation. |
|
|
Constant-Force
Pulling on
a
Lennard-Jones Polymer
Patrick
O’Neill
The end-to-end distance of a linear Lennard-Jones polymer is examined,
with one end fixed to a surface, and a constant pulling force applied
to the other end. The polymer is initially in a coiled state, and
uncoils with sufficient pulling force. The average uncoiling rate
depends on the pulling force, and for insufficient pulling force, the
polymer remains in the coiled state for long times. |
|

|
Modeling
the Fullerene
Distribution in BHJ Solar Cells
James
T.
Rogers
A simple model was used to study the effect of polymer molecular weight
on the distribution of fullerene molecules within a bulk heterojunction
(BHJ) solar cell. Measurements of the pair distribution function (PDF)
derived from this model indicate improved local structural ordering of
the fullerene molecules with increasing molecular weight. This is well
correlated with recent experimental results which have shown drastic
increases in solar cell performance with increasing polymer molecular
weight. Further development of this model should allow identification
of polymer/fullerene chemistries which may result in even more optimal
blend morphologies. |
|
|
A
Coarse-grained MD Simulation of Micellization Process
Mansi
Seth
A Molecular Dynamics Simulation was performed to study the process of
Micellization of surfactant molecules in water, using an implicit
solvent, coarse-grained model. The Critical Micelle Concentration
(C.M.C) was determined for an ionic surfactant. The Effect of
Temperature on C.M.C was also studied. Micellization is observed only
above a certain critical concentration, after which there is an
increase in concentration of micelles, with increase in surfactant
concentration. An increase in temperature leads to increase the C.M.C
of the surfactant. The results obtained are thus, in qualitative
agreement with experimental observations and those from other
simulations of ionic surfactant molecules. |
|
|
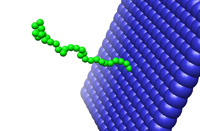
Molecular
Dynamics of a
Tethered
Protein
Brad
Spatola
The system being investigated is the interaction between a bound
protein molecule, modeled as 30 Lennard-Jones particles, and a
surface. In order to examine the protein folding into the
wall, the radius of gyration and protein end-to-end length were
calculated and a molecular dynamics movie was recorded.
Results indicate that the protein will fold into a tighter group at
lower temperatures than at higher temperatures. In addition,
once the protein approaches the wall, it will stay in a closely packed
formation for the duration of the simulation run. |
|
|
|